A More Complex Future Market for Diagnostics?
Summary
With regulatory changes on the horizon, diagnostics stakeholders should prepare for a new evidence development and commercialization environment.Defining Diagnostics
Several categories of diagnostics are defined by regulators such as the Food and Drug Administration (FDA) and Centers for Medicare and Medicaid Services (CMS), among others. Specifically, in-vitro diagnostic tests (IVDTs) are subject to FDA enforcement both pre- and post-market, while laboratory developed tests (LDTs) are IVDTs that are manufactured by and used within a single laboratory and under validation purview of CMS within the Clinical Laboratory Improvement Amendments (CLIA).
These distinct regulatory frameworks for IVDTs and LDTs can create a non-standardized market, resulting in confusion around expectations for evidence generation and go-to-market commercialization strategies. The FDA is expected to issue notable regulatory changes to LDTs this spring, prompting developers to reexamine their market strategies.
Current Regulatory and Commercialization Market
Diagnostic developers are advised to consider several aspects when commercializing diagnostics, each of which has unique strategic implications depending on whether it is an IVDT or LDT under existing regulatory and market frameworks:
- FDA Regulations: Diagnostics are currently regulated within the medical device statutory framework. While IVDTs typically require FDA clearance/approval, the agency is currently applying regulatory discretion with respect to LDTs, as they are not explicitly mentioned in statute. The FDA’s final rule on LDTs is anticipated to change this.
- CMS and American Medical Association (AMA): Developers need to ensure that their diagnostic products and corresponding services can be ordered in the market via appropriate codes that facilitate utilization, payment, and reimbursement through payer claims management. Most payers would require the use of codes maintained and generated under CMS and AMA for reimbursement, such that a diagnostic’s broad access to providers and patients is highly dependent on this step.
- Commercial Payers: Once developers secure codes for their diagnostic, they need to ensure that payers deem the product reasonable and necessary for clinical care, leading to medical policy coverage for the intended use of the diagnostic. If covered under the commercial payer medical policy, the diagnostic services reimbursement rates are often negotiated to the payer’s rates as a percentage of the CMS Clinical Laboratory Fee Schedule rates.
- Molecular Diagnostic Program (MolDx)/Medicare Administrative Contractors (MACs): Select jurisdictions under individual MACs have established specific programs for evidence related to coverage and payment of molecular diagnostics. Within the MolDx program, developers must obtain Diagnostics Exchange Z-Codes as a test-specific identifier, in addition to an AMA-assigned Current Procedural Training (CPT) code, to help track utilization of a specific molecular diagnostic test. Neither coding system guarantees coverage and payment, but are necessary components for reimbursement.
Regardless of the forthcoming FDA final rule regarding regulatory oversight of LDTs, diagnostic developers will need to consider a variety of steps to obtain market access, making it imperative to consider a short, medium, and long-term strategic horizon for commercialization (Figure 1).
Figure 1. The Diagnostics Market Is Complex, With Multiple Access Influencers and Evolving Dynamics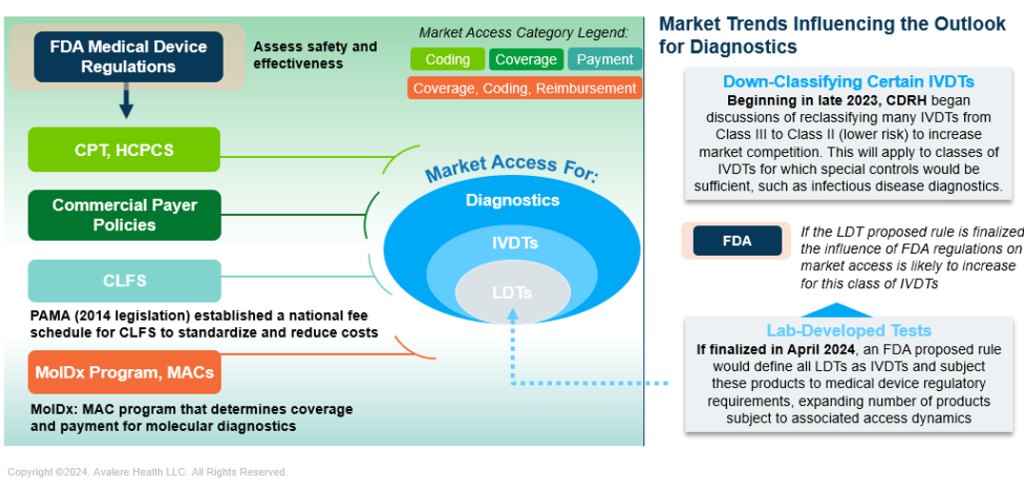
CDRH: Center for Devices and Radiological Health; CLFS: Clinical Laboratory Fee Schedule; CPT: Current Procedural Terminology; HCPCS: Healthcare Common Procedure Coding System; MAC: Medicare Administrative Contractor; MolDx: Molecular Diagnostics; PAMA: Protecting Access to Medicare Act
Forthcoming FDA Regulatory Changes
This spring the FDA is expected to finalize a proposed rule on LDTs that would explicitly define LDTs as a type of IVDTs and thus subject them to medical device regulation. Separately, FDA has announced plans to reclassify many infectious disease IVDTs, from Class III to Class II. The FDA has stated that Class II controls are sufficient to ensure safety and effectiveness while increasing competition in this area due to a lower burden of proof.
The LDT rule is particularly paradigm changing. As most LDTs are not currently subject to FDA regulations, this will create new strategic burdens on laboratories and partners developing these tests. Since FDA clearance/approval will be one of the first requirements in the lifecycle of an LDT, this may have downstream implications for commercialization.
Implications for Diagnostic Manufacturers and Laboratories
Stakeholders in the industry, particularly diagnostic manufacturers, will need to consider adapting current development and commercialization strategies in response to anticipated policy changes. Others, such as pharmaceutical sponsors with therapeutics that rely on biomarker identification/companion diagnostics, will benefit from considering the overall changing diagnostic landscape as part of their partnership strategies. Considerations may include:
- Reevaluating currently marketed and pipeline diagnostics with respect to FDA requirements for safety and effectiveness to better understand new evidence requirements;
- Reassessing prioritization of products based on go-to-market evidence burden relative to the market potential;
- Establishing and expanding evidence generation plans as necessary, including developing health economics and outcomes research roadmaps;
- Capitalizing on the value of real-world data for on-market diagnostics (e.g., targeted evidence by population and intended use);
- Assessing development and/or commercialization opportunities that may arise with new targeted evidence (this may include evidence supporting use of products in diverse patient populations to facilitate equitable access);
- Considering collaboration opportunities between pharmaceutical and diagnostic sponsors to enhance treatment access.
A silver lining may be that with a clear FDA mandate over LDTs, stakeholders may see greater certainty with respect to evidence requirements and opportunities for marketing clinical claims in the future.
Next Steps
Avalere experts in regulatory, evidence, and market access strategy can help you navigate these evolving dynamics for LDTs, including potential regulatory changes in transition and the broader impact of these policies on patients, payers, and access. To learn more about how we can help your organization prepare and respond to the changes, connect with us.
Related Content
