
Regulatory Strategy and FDA Policy
The development of drugs, biologics, and medical devices follows rigorous paths to ensure safe and effective medicines. We create FDA regulatory engagement strategies and help clients interpret FDA regulations, guidance, and the likely impact of legislation. Interpreting FDA priorities, resources, User Fee Acts, and label and promotion requirements, our experts can help you manage your pipeline.



FDA Reform: It’s Time to Act, But Not as an Independent Agency
As former federal officials, Dan Troy, David Beier, and I share our perspectives on the call by former FDA officials representing both political parties to make the FDA an independent agency separate from its parent, HHS.
Landscape for Diagnostics Will Continue to Change in 2019
While precision medicine is expected to revolutionize patient therapy, the increasing complexity of diagnostics is leading policymakers to revamp the way these tests are regulated and paid for.

FDA Seeks Stakeholder Input on How to Enhance Biosimilars and Interchangeable Biologics Market
Earlier this week, the Food & Drug Administration (FDA) released a Part 15 public hearing announcement and request for comment on how the FDA can facilitate greater availability while balancing competition and innovation for all biologics.

ICER’s Use of FDA Approval Volume to Calculate Budget Impact Thresholds: A Scenario Analysis
ICER’s reliance on the average number of FDA drug approvals to calculate budget impact thresholds leads to significant variability.
Interview: E3 – Real-World Evidence: Is It a Game Changer for the FDA?
We discuss 3 steps companies should complete now to take advantage of the FDA’s priority to advance the adoption of RWE to support agency decision making.

Use of Step Through Policies for Competitive Biologics Among Commercial US Insurers
Avalere evaluated payer policies for biologics when biosimilars are available.

Two-Year Countdown Begins for FDA “Roll-Over” of Biologics Currently Regulated as Drugs
In just two years, on March 23, 2020, biologics currently regulated as drugs will transition to being regulated as biologics. Many aspects of how FDA will implement this transition have yet to be established.
Issue Brief: The Elements of a Competitive Biologics Market
Most people are familiar with generic drugs as less costly alternatives to drugs whose patents have expired.

Three Ways the FDA Can Facilitate Claims of Interchangeability
In the US, an interchangeability designation by the Food & Drug Administration (FDA) is perceived as the holy grail in biosimilars development by some and yet regarded as irrelevant by others.
Interview: E1 – Real-World Evidence: Is It a Game Changer for the FDA?
In our real-world evidence series, Avalere experts explore real-world evidence as a disruptive force in healthcare. In Episode 1, we focus on why RWE is such a hot topic.
FDA Prepares for the Next Generation of Regenerative Medicines
In its new regulatory framework for regenerative medicines released in November 2017, FDA takes a flexible approach, creating opportunity for manufacturers.

FDARA Builds on Progress in Expanded Access
The FDA Reauthorization Act of 2017 (FDARA), which was signed into law on August 18, 2017, calls on FDA to examine underlying barriers to access to investigational drugs.

Our Take on the Senate Passage of the FDA User Fee Reauthorization Bill
Today, the Senate voted to pass H.R.2430, the FDA Reauthorization Act of 2017.
Avalere Deepens Focus on FDA Regulatory Matters with Three New Hires
Tom Kraus, who most recently served as the chief of staff at the FDA, will be joining Avalere as senior vice president.
Five Obstacles to Competition in the United States Biologics Market
Biosimilars have the opportunity to foster competition, but policy and market barriers limit the growth of a functioning market.
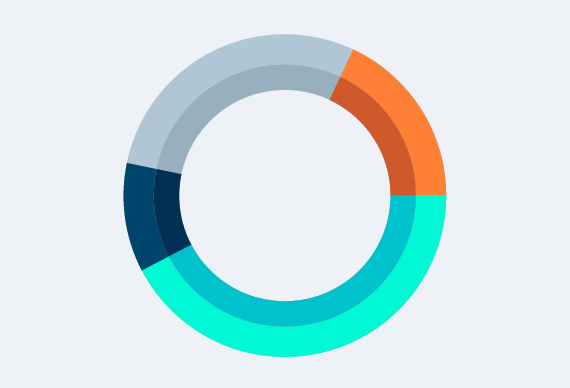
Manufacturer’s Compassionate Use Policies: Companies with Posted Policies More Than Doubled Since September 2016
The 21st Century Cures Act requirement to post compassionate use policies may explain increase.
FDA Priority Review Voucher Programs: Incentive for Industry to Develop Novel Treatments
Extension of the Sunset Clause provides additional opportunity for the FDA to award vouchers and for transference of vouchers from recipients to other sponsors.
Interview: E2 – Highlights of FDA and NIH in 21st Century Cures
The final signing of the 21st Century Cures Act is the culmination of 3 years of efforts by lawmakers in the House and Senate to expedite developing and making available new treatment options. In the final episode in our series, Jay Jackson discusses what changes are on the horizon for products under The Cures Act.

FDA Has Received $7.67 Billion from Manufactures to Fund Drug Review
Since 1992, the Food and Drug Administration (FDA) has collected $7.67 billion in user fees from pharmaceutical manufacturers to fund drug reviews based on an Avalere analysis of FDA data.
Interview: An Overview of FDA’s Expanded Access Guidances
Our expert, Jay Jackson, discusses recent final guidance issued by the FDA on compassionate use.